



Prof. Yousung Jung’s group at KAIST has taken a major step toward a dramatically accelerated material design from the conventional, costly, all DFT-based high throughput screening approaches by adapting a machine learning algorithm and uncertainty-based screening framework. The proposed approach could be a fast alternative to accelerate the exploration of demanding materials space in various applications. The study was published on March 25 in Journal of Chemical Information and Modeling (Uncertainty-Quantified Hybrid Machine Learning/Density Functional Theory High Throughput Screening Method for Crystals, J. Chem. Inf. Model., 60, 1996-2003 (2020)).
The advances in the fundamental understanding of crystal structures have accelerated the discovery of new materials for renewable and sustainable energy technologies. Previous efforts on new materials design have largely depended on chemical intuition learned from the prior knowledge, but the latter strategies alone can often be biased to incremental improvements of the existing materials, rather than exhaustively searching the unseen chemical space combinatorially. In this regard, the first-principles-based theoretical approaches (specifically, computational high-throughput screening, HTS) has been a powerful tool to expand the search space to discover new materials in the past decades. However, first-principles density functional theory (DFT) does not scale favorably with the system size, O(N3), and thus a large number of DFT calculations needed for HTS have been the major bottleneck for an exhaustive exploration of the chemical space.
To overcome this challenge, other researchers had previously proposed machine learning (ML) approaches as an alternative to DFT since it rapidly predicts the property of crystals using the data-driven knowledge leveraged by the materials database. Especially, novel graph-based ML models have been recently introduced with significantly improved accuracy which encodes crystal structural information to a single vector during the learning structure−property relation. However, one of the open challenges of such an approach is that the force cannot be evaluated in the present form, and hence if the energy of the given structures is largely changed after DFT relaxation, the reliability of prediction results on the DFT-unrelaxed initial substituted geometry would be low. Therefore, addressing the uncertainty caused by the use of non-relaxed geometry remains a critical limitation of the current substitution-based HTS approaches.
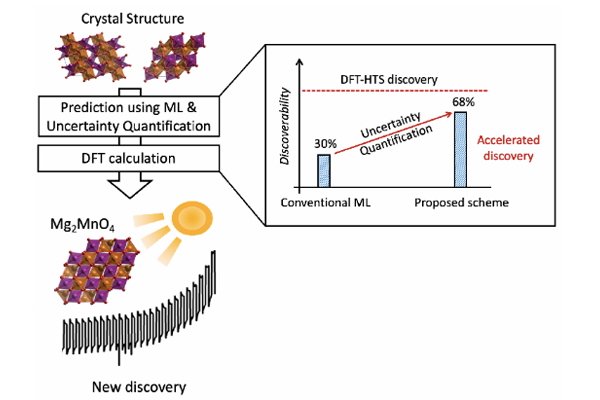
The researchers assess the usage of a graph-based ML as an alternative to DFT-based HTS, and attempt to address the prediction uncertainty caused by the deformation in the crystal structures using the dropout-based Bayesian modeling. The model is trained with ~70,000 inorganic crystals taken from Materials Project open database, and used to explore >7,000 Mg-Mn-O hypothetical crystal structures for comparison to previous brute-force all DFT-based HTS scheme. The proposed graph-based ML and dropout-based Bayesian modeling show significant improvement of the screening performance (discoverability), by more than a factor of 2 compared to the conventional ML-based HTS approach. Notably, the present ML/DFT hybrid HTS scheme showed the enhanced screening efficiency by more than a factor of >50 with the 68% discoverability compared to the conventional DFT-based HTS method.
“The current uncertainty-quantified machine learning approach can be used in many practical materials design applications that require numerous costly first principles calculations in high-throughput screening”, said Yousung Jung, professor of chemical and biomolecular engineering at KAIST who co-authored the paper.
This work was supported by the Saudi-Aramco-KAIST CO2 Management Center, and by supercomputing time from the Korea Institute of Science and Technology Information.
Mr. Juhwan Noh, Prof. Yousung Jung Dept. of Chemical and Biomolecular Engineering, KAIST
Homepage: http://qchem.kaist.ac.kr
E-mail: ysjn@kaist.ac.kr
